


R&D
Mission drives innovation, and innovation improves quality of life
Medical Information
[In-depth Report] BCR-ABL1 allosteric inhibitor symbolizes a new start oftargeted therapy
2021-10-15 Views:
Allosteric regulation is ubiquitous in the life processes of cells, including non-covalent interactions between molecules, covalent modifications (phosphorylation, point mutations, and small-molecule chemical reactions), cellular environment (temperature, radiation, pH and ionic strength) and light absorption.[1–4]. Allosteric effects precisely control the normal life processes of cells, including cell signal transduction, metabolism, enzymatic catalysis and gene regulation. Disorders of protein allosteric regulation are closely related to many diseases, such as cancer, mental disorders, diabetes, inflammation, and immune diseases [5-7].
Allosteric drugs refer to the effector molecules that regulate the biological functions of proteins by binding to allosteric sites (spatially different from normal active sites or substrate binding sites) on the target proteins [5,8,9]. Such new modulators may affect the biological effects of proteins in two ways. One is that allosteric regulation triggers the perturbation and reorientation of the allosteric site and its neighboring atoms, thereby generating strain energy and forcing the next layer of atoms to move, thus affecting the next layer of atoms. Allosteric perturbation propagates from allosteric site to orthosteric site through allosteric waveforms, resulting in fine-tuning of the conformational dynamics of orthosteric site [1,6,10]. The other is the recently proposed kinetic-driven allosteric concept. Allosteric perturbations spread throughout the entire protein structure (including orthosteric sites), which changes the vibration mode and conformational population of the entire protein, and can trigger major changes in protein conformation [11,12].
(I). Allosteric modulators - an emerging trend in R&D
Compared with traditional orthosteric drugs, allosteric drugs have several significant advantages: 1) Overcoming drug resistance: in antibacterial, antiviral and antitumor treatments, drug resistance has always been a problem, while allosteric modulators and orthosteric modulators act on different sites of the target to exert pharmacological effects based on different mechanisms of action, thus the allosteric modulators have the potential to overcome the acquired resistance emerging upon treatment with the orthosteric modulators [13]; 2) Presenting high selectivity: the sequence homology of the allosteric site is low, and the amino acid residues are low in conservation, resulting in higher selectivity, lower off-target toxicity and lower-dose prescription [14]; 3) Drugging the undruggable targets: some residues in binding pockets at orthosteric sites are highly conserved, and are polar and charged, which not only lead to weak selectivity and high toxicity of the modulator, but also the poor cell permeability and oral bioavailability of the modulator and lack of druggability; secondly, the surface of the orthosteric sites is large and flat, lacking suitable small molecule binding pockets (such as protein interaction interface targets); or the natural substrate (ATP/GTP) possesses very high concentration in the cells, and has very strong affinity with the active site, making it extremely difficult for the modulator that directly target the orthosteric site, etc. [15-16]; 4) Allosteric regulation offering potential for more precise regulation: some proteins have limited functional windows, which require precise regulation. Allosteric modulators can regulate protein function in two directions, thus playing a role of precise modulation. For example, allosteric agonists are less likely to induce protein desensitization after activation [17].
Since allosteric modulators show great development prospects and their significant advantages over traditional orthosteric drugs, the target allosteric effect has been established as a new mechanism for drug discovery, triggering an upsurge in allosteric drug discovery, optimization, and clinical development, attracting many biotechnology companies and large pharmaceutical companies to flood into this focused field, promoting many large-scale financing, cooperation and transactions [18]. Below, we will take the BCR-ABL1 allosteric inhibitor as an example to comprehensively introduce the development history, mechanism of action, clinical progress, application potential and other aspects of this new type of drug.
(II). Development history of BCR-ABL1 inhibitors
Imatinib, as the first targeted therapy drug on the market, opened up the era of small-molecule targeted therapy for tumors, and was hailed as a silver bullet by the Time magazine (USA). The successful development of this BCR-ABL1 inhibitor turned chronic myelogenous leukemia (CML), which had a high fatality rate, into a chronic disease, and the treatment of CML has thus achieved milestone progress [19]. However, 10-20% of CML patients developed acquired resistance after treatment with imatinib, mainly because the BCR-ABL1 gene can generate more than 50 point mutations [20]. Subsequently, several second-generation drugs were marketed, which revolutionized the treatment regimens of CML and brought more clinical benefits to imatinib-resistant CML patients, but most patients still need to continue medication. In addition, the most challenging and most common resistance mutation BCR-ABL1T315I confers resistance to all available first- and second-generation BCR-ABL1 inhibitors. Although ponatinib, which is marketed abroad, can overcome BCR-ABL1T315I resistance, it can cause treatment-related risks of arterial thrombosis and liver toxicity. Also, the results of in vitro experiments have shown that some rare single-point mutations and compound mutations can also confer resistance to ponatinib [21]. At present, many domestic and foreign pharmaceutical companies are still actively developing the third-generation BCR-ABL1 inhibitors. However, these inhibitors are still orthosteric inhibitors that compete with ATP for active sites, and their structure is similar to that of ponatinib. Therefore, the similar safety and cross resistance problems as those of ponatinib still exist.
In order to overcome the resistance conferred by the above-mentioned BCR-ABL1T315I and single-point or compound mutations to ponatinib, and to solve the intolerance and toxicity problems of existing drugs, the pharmaceutical giant Novartis took the lead in deploying the fourth-generation BCR-ABL1 allosteric inhibitor (ABL001), and the project is currently undergoing phase III clinical study; domestically, the fastest progress is made on TGRX-678 developed by Shenzhen TargetRx, Inc. (hereinafter referred to as TargetRx). This BCR-ABL1 allosteric inhibitor has entered the clinical development stage.
(III). The mechanism of action of BCR-ABL1 allosteric inhibitors
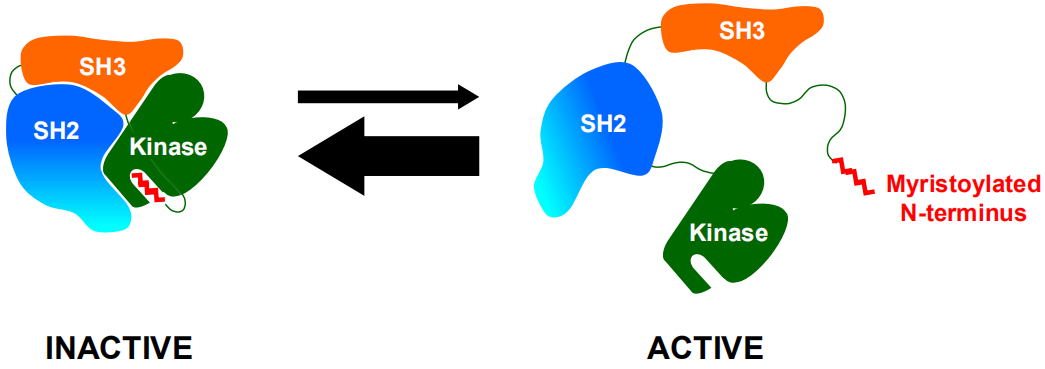
Under normal circumstances, the allosteric site of the ABL1 kinase domain is occupied by its own N-terminal myristoyl peptide, which induces cross-linking of the SH3-SH2-kinase domain to play a key role in negatively regulating ABL1 kinase activity, and regulates the transduction of cell normal proliferation signals (Figure 1). When BCR is fused with the ABL1 gene, the N-terminal myristoyl peptide of BCR-ABL1 kinase is lost, resulting in the vacancy of the allosteric site on the ABL1 kinase domain, then the SH3-SH2-kinase domain cannot be induced to cross-link, and the self-inhibition balance is broken to be an activated open conformation, which causes continuous activation of BCR-ABL1 kinase, and induces cell proliferation and tumor formation (Figure 2) [22,23].
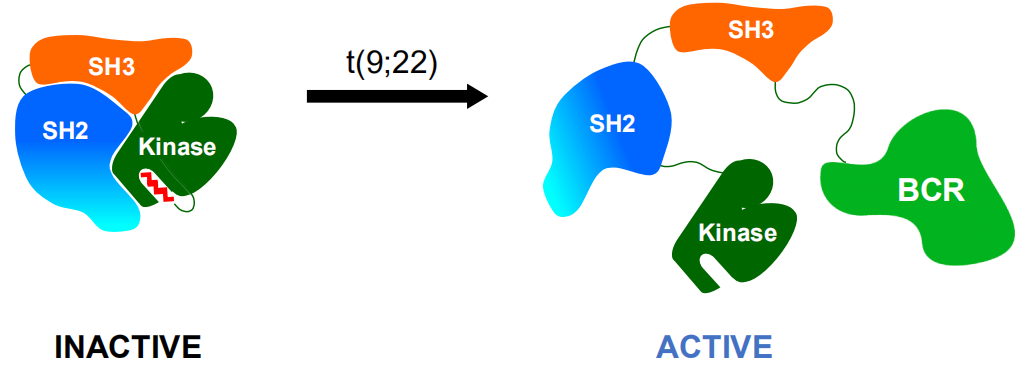
In order to restore the negative regulatory function of ABL1 kinase, Novartis and TargetRx developed ABL001 and TGRX-678 respectively by mimicking the function of the myristoyl N-terminal peptide of natural ABL1 kinase. These two molecules bind to the myristoyl vacancy allosteric site at the C-terminus of the kinase domain (Figure 3a, purple spherical region), so that the SH3 and SH2 domains are cross-linked to the kinase domain, the ABL1 kinase is changed from the active carcinogenic state to closed inactivated non-carcinogenic state, and the self-inhibitory conformation of ABL1 kinase is re-stabilized (Figure 3), thereby exerting an antitumor effect [24].
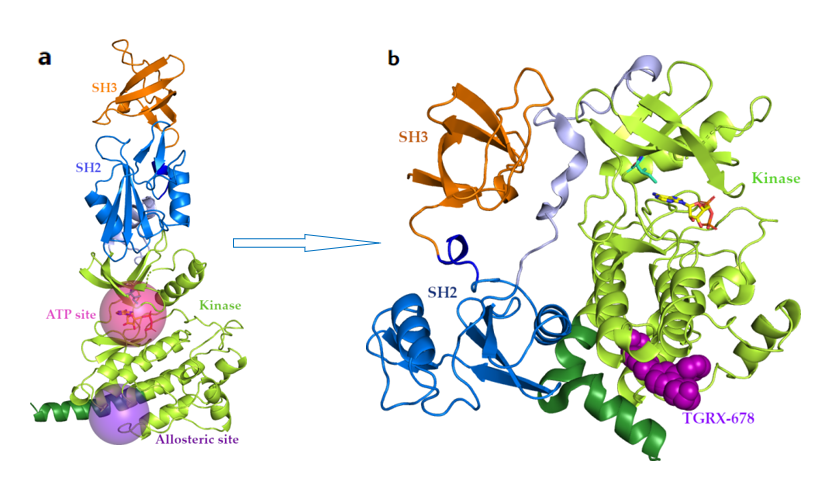
It can be seen that the functional site and mechanism of action of the fourth-generation BCR-ABL1 inhibitors are completely different from those of the previous three-generation orthosteric inhibitors. The molecular mechanism of the first-generation inhibitor imatinib, the second-generation inhibitors dasatinib, nilotinib, bosutinib, radotinib and flumatinib, and the third-generation inhibitor penatinib is to bind to the "hinge region" of the ATP binding site (Figure 3a, red spherical region), which are ATP competitive inhibitors that inhibit the autophosphorylation and substrate phosphorylation of BCR-ABL1 kinase, thereby inhibiting the proliferation of cancer cells and tumor formation. They are all typical orthosteric inhibitors. ABL001 and TGRX-678 act on the allosteric site of the BCR-ABL1 kinase domain, which is located at the C-terminal myristoyl binding site of the ABL1 kinase catalytic domain (Figure 3a, purple spherical region), far away from the orthosteric ATP active site and T315I mutate amino acid residues, so that the binding of ABL001 and TGRX-678 to BCR-ABL1 is almost unaffected by T315I mutation.
The allosteric regulation of ABL001 and TGRX-678 can trigger the fine-tuning of the conformational dynamics of the orthosteric site in the form of allosteric waves, so that nilotinib, which is ineffective against resistance mutation T315I, can re-bind to the orthosteric site of the BCR-ABL1T315I kinase to restore its inhibitory effect. In addition, the allosteric effect driven by the kinetics of allosteric inhibitor causes the SH3 and SH2 domains to cross-link to the kinase domain, resulting in major conformational changes in the entire protein, so as to form the inactivated non-carcinogenic state. Although BCR-ABL1Y253H/T315I compound mutations are resistant to ponatinib, some studies have suggested that when combined with ABL001, ponatinib can temporarily occupy the orthosteric active site to induce conformational changes from the DFG-in active conformation to the DFG-out inactive conformation. This conformational change greatly reduces the flexibility of the myristoyl allosteric site, allowing it to assemble and better bind to ABL001. Conversely, the binding of ABL001 to the allosteric site is expected to result in a further continuous and stable binding of ponatinib and the DFG-out inactive conformation of BCR-ABL1 kinase. This kind of orthosteric-allosteric coordinated regulation mechanism of action ultimately leads to strong kinase inhibition to overcome mutation-induced resistance to ponatinib [21]. Therefore, the combination of orthosteric inhibitors and allosteric inhibitors can exert a strong synergistic effect and produce unexpected therapeutic effects.
(IV). Results of preclinical and clinical trials of BCR-ABL inhibitors
Pre-clinical studies have shown that ABL001 of above 0.1nM has the same inhibitory rate on kinases regardless of the low concentration (10 uM) or high concentration (2 mM) of ATP, indicating that ABL001 is an effective BCR-ABL1 non-ATP competitive kinase inhibitor. ABL001 has strong biological activity on BCR-ABL1 wild-type, T315I and other mutant Ba/F3 cells (Figure 4), and can effectively address the problem of acquired resistance emerging upon treatment with orthosteric inhibitors, especially the resistance caused by the mutations at the gatekeeper residue T315I of the first and the second-generation inhibitors [24].
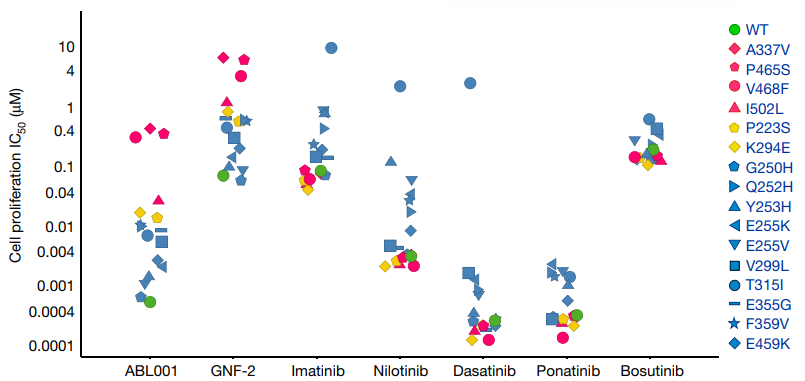
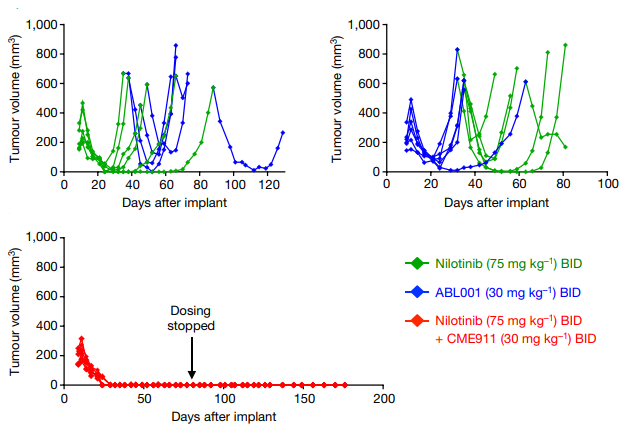
In addition, a variety of experiments have evaluated the selectivity of ABL001. ABL001 at a concentration of less than 10 uM may not inhibit the phosphorylation of more than 60 other recombinant kinases [24]. According to the evaluation of the activity and selectivity of ABL001 allosteric inhibitor and orthosteric inhibitor using 450 lines of tumor cells, ABL001 showed excellent anti-proliferative activity (IC50=1-20 nM) against BCR-ABL1 positive tumor cells. But inhibitory activity is low against BCR-ABL1 negative tumor cells (IC50>1000 nM), and its selectivity is better than that of orthosteric inhibitors, as shown in Figure 5. In addition, ABL001 at a concentration of <3 uM has no substantial effect on G protein-coupled receptors, cell transporters, ion channels, nuclear receptors and enzymes [25]. ABL001 not only overcomes the resistance to the first and second-generation inhibitors, but also highlights its high selectivity, which may avoid the serious side effects caused by the third-generation inhibitor pentatinib. Preclinical studies have also shown that the combination of ABL001 and nilotinib achieves a strong therapeutic effect, and can completely control the tumor progression of xenograft tumor mice, and the disease is not easy to relapse after stopping medication (Figure 6) [24]. The combination of ABL001 and ponatinib can not only produce activity against compound mutations Y253H/T315I, E255V/T315I, etc., which are most resistant to ponatinib, but also effectively inhibit the occurrence of compound mutations in BCR-ABL1 and reverse ponatinib resistance, which systematically solves the problem of drug resistance caused by compound mutations, is expected to greatly reduce the concentration of penatinib required in clinical practice so as to reduce side effects, and provides an effective treatment strategy for CML patients [21].
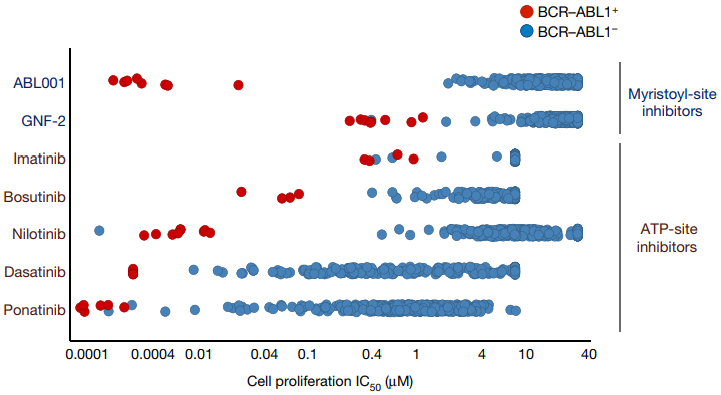
Clinical studies have shown that CML patients who are resistant to previous orthosteric inhibitors or have unacceptable side effects and have received at least two orthosteric kinase inhibitors, including those who have failed in ponatinib treatment and those with T315I mutations, ABL001 showed good safety and effectiveness [26]. The clinical trial of the combined application of ABL001 and imatinib has shown promising initial efficacy, and this combination has good safety and tolerability [27]. The results of a phase III clinical trial ASCEMBL of ABL001 showed that compared with bosutinib, the 24-week major molecular remission (MMR) rate was more significant and better, and the study reached the primary endpoint [28]. Researchers are evaluating ABL001 in combination with other orthosteric kinase inhibitors as a first-line treatment for CML. ABL001 has achieved initial results in clinical trials for the treatment of CML, indicating significant clinical medicinal value.
TGRX-678 is the first and fastest-developing fourth-generation BCR-ABL1 allosteric inhibitor in China, and the second BCR-ABL1 allosteric inhibitor in the world. It has obtained clinical approval in China (approval number: CXHL2000158/CXHL2000159), and clinical trials are actively prepared. The results of preclinical in vivo and in vitro studies show that compared with ABL001, TGRX-678 has higher activity and selectivity for Ba/F3 Bcr-AblT315I cells, better oral bioavailability, and better safety in animals, and has the potential to become a best-in-class, so its clinical performance is expecting.
(V). Summary and Prospects
Based on the novel site of action and mechanism, BCR-ABL1 allosteric inhibitors can be used to treat CML patients who were resistant or intolerant to orthosteric inhibitors (including drug resistance due to the T315I gatekeeper residue mutation). The combination with orthosteric inhibitors can also overcome resistance resulting from single point and compound mutations to ponatinib, reduce the occurrence of drug resistance, reverse the acquired drug resistance, reduce the dose, and relieve toxic and side effects. The patients are expected to keep treatment free remission (TFR) for a long time, achieving functional cure. BCR-ABL1 allosteric inhibitors are also expected to be used in the treatment of other tumors or diseases, greatly expanding its clinical indications and medicinal value. It is hoped that these new types of drugs can open a new chapter in targeted therapy and guide new directions for the development of more drugs that overcome drug resistance.
References:
Prev: Last

 Tel: +86-0755-86934300
Tel: +86-0755-86934300 3rd Floor, Building A1, Kexing Science Park, No. 15 Keyuan Rd., Nanshan District, Shenzhen, China
3rd Floor, Building A1, Kexing Science Park, No. 15 Keyuan Rd., Nanshan District, Shenzhen, China E-mail:
E-mail: 